 |
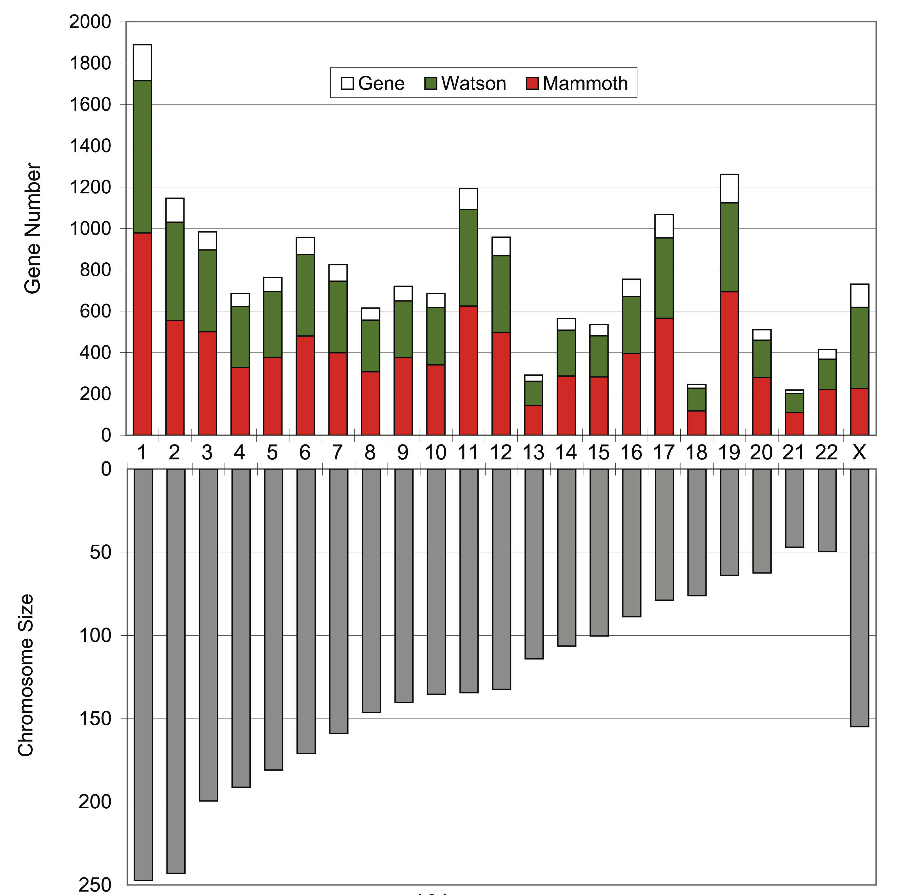
How much of the woolly mammoth genome did we sequence? Since typical genome sizes of placental mammals are around 3 Gb (1 Gb = 1 billion nucleotides of DNA), one might expect that our 3.3 Gb of mammoth sequences would cover most of the mammoth genome. However, the African elephant genome has previously been estimated at between 4.2 and 4.8 Gb using an experimental method called the C-Value technique (Redi et al. 2007), which, although less accurate than genome sequencing, has consistently predicted the Afrotherian genomes to be larger than previously sequenced placental genomes. We estimated how much of the mammoth genome has been sequenced by searching for matches to a set of predicted elephant genes that could be confidently mapped to unique positions on human chromosomes (see the figure below). We also searched for the so-called ultraconserved regions defined by Bejerano et al. 2004, which are intervals of at least 200 consecutive nucleotides that are identical among human, mice, and rats, and hence very likely to be found in all mammals. In both cases, around 50% of the bases were found, where some of them matched more than one mammoth read. Accounting for multiple reads that include the same genomic position, this translates into 0.7-fold coverage, i.e., that the total length of our mammoth reads is 70% of the genome's true length. Since some of our reads are very short, and hence difficult to align reliably, this may well be an under-estimate. This difference between 50% and 70% can be a bit confusing at first, and perhaps the following analogy will help to clarify things. Suppose you throw 70 darts at a dart-board that has 100 positions. The number of positions that are hit can vary from 1 (if all darts land on the same position) to 70 (if different darts never land in the same position). With random throws, statistically we expect about 50 positions to be hit (some of them multiple times). In general, if the number of darts is 70% of the number of positions, then on average 50% of the positions will be hit. The essence of our problem was that we could measure the fraction of positions that were hit and wanted to estimate the ratio of darts to positions. The following figure indicates, for each human chromosome except Y, what fraction of the gene-encoding bases on that chromosome were found in our mammoth sequences. For most chromosomes it was close to 50%. For chromosome X the fraction is much lower because most of our data came from M4, which was a male. For comparison, we show (in green) the fraction that our method finds for the genome sequence of Dr. James Watson (see Wheeler et al. 2008). |
 |
| References
Bejerano, G. et al. Ultraconserved elements in the human genome.
Science 304, 1321–1325 (2004). |
 |